An automated workflow for high throughput free energy perturbation (FEP) simulations for ligand series.
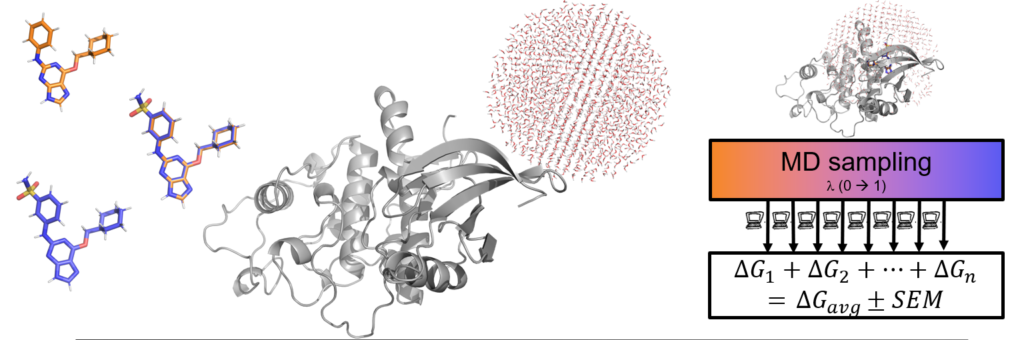
QligFEP is based on a dual topology approach, which defines a univocal perturbation pathway between a given ligand pair. Within the same framework, one can perform scaffold hopping, linker length changing and pose ranking.
Large ligand series can be processed thanks to our proprietary technology, QmapFEP, which optimizes the definition of the alchemical transformations needed to cover the whole series.
QligFEP is flexible, and allows the usage of different forcefields and sampling options.
Fast and accurate, thanks to the spherical boundary MD simulations within the Q package. Read more about the large benchmark of QligFEP here.
QligFEP: an automated workflow for small molecule free energy calculations in Q
W. Jespers, M. Esguerra, J. Åqvist, H. Gutiérrez-de-Terán, Journal of Cheminformatics 2019, 11 (1), 1-16.