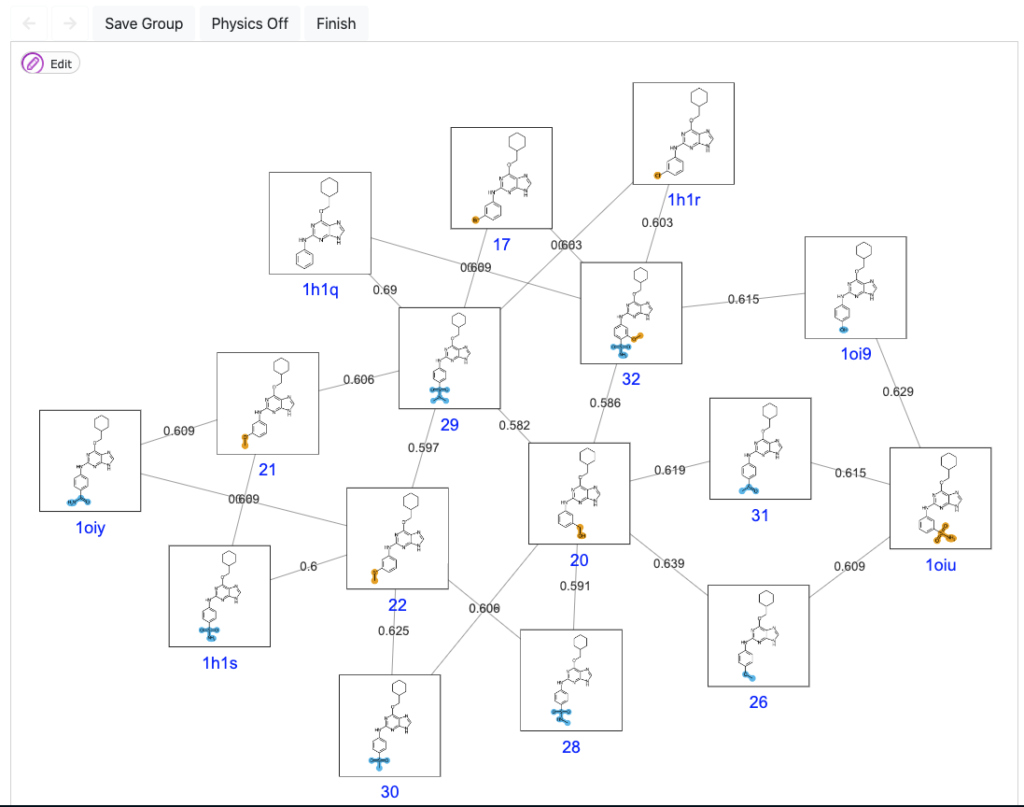
An optimized path through chemical space.
When designing a FEP campaign for ligand optimization, it is key to have a robust and automated method to map the ligand series into a number of FEP pair comparisons. The main goal is to obtain the best trade between optimal computational yields and highest predictive accuracy. In ModSim Pharma, we have solved this issue with the development of our own algorithm, QmapFEP.
QmapFEP is based on Jarnik’s algorithm for finding the minimum spanning tree for a weighted undirected graph, but optimized for the case of ligand datasets which are described with Morgan chemical fingerprints. The graph is designed to allow cycle closure correction to estimate errors and, ultimately, absolute binding affinities taking as reference compounds with known experimental affinity.
Our algorithm has consistently shown a ratio of 1.5 nodes per ligand, which is way beyond other FEP mapping applications, contributing to a higher efficacy while maintaining the accuracy. You can read more on the performance of QligFEP with QmapFEP here.